

I类医疗器械
I 类医疗器械可以根据 MDR 自行声明符合 CE。自我声明意味着既不需要公告机构认证,也不需要任何认证机构的任何其他类型的批准!I 级的风险最低。在这种情况下,制造商可以对其进行自我符合性声明即“自我认证”。
如何进行I类医疗器械符合性声明
以下是在 I 类医疗器械上加贴 CE 标志的步骤:
- 根据附件 II 和 III 准备技术文件
- 符合性声明按照附件 IV 准备
- 根据附件 V 加贴 CE 标志
Is 类医疗器械(无菌医疗器械)
I类医疗器械具有低/中等风险。在这种情况下,以无菌状态投放市场的器械,公告机构的参与仅限于与确保和保持无菌状态有关的制造方面。
Im 级(测量设备)
Im 类医疗器械具有低/中等风险。在这种具有测量功能的设备投放市场的情况下,公告机构的参与仅限于与设备符合计量要求有关的制造方面。
Ir 类医疗器械(可重复使用的手术器械)
Ir 类医疗器械具有低/中等风险。在这种可重复使用的手术器械投放市场的情况下,公告机构的参与仅限于与器械重复使用相关的方面,特别是清洁、消毒、灭菌、维护和功能测试以及相关的使用说明。
CE符合性评估路线
对于Class Is / Im / Ir 医疗器械(检验仅限于无菌/计量/重复使用,如适用),合格评定路线如下:
根据附件 II 和 III 准备技术文件(技术文件)
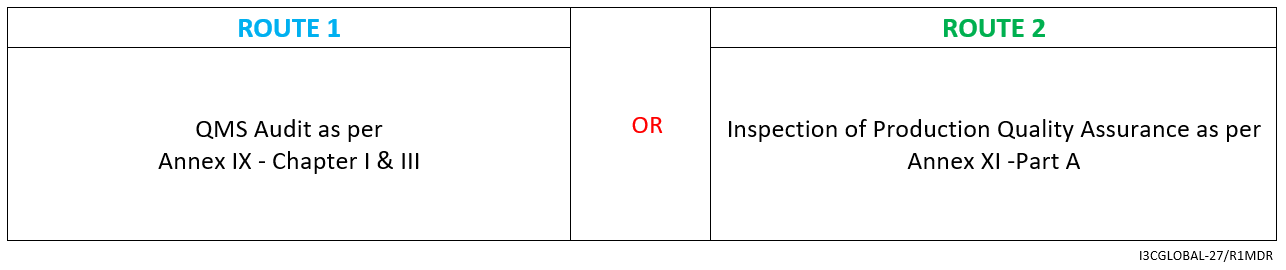
- 符合附件 IV 的声明
- 根据附件 V 加贴 CE 标志
将 I 类医疗器械投放欧盟市场的步骤
- 确认产品为医疗器械
- 确认产品为 Is 类或 Im 或 Ir 类医疗设备
- 确认是否满足一般安全和性能要求
- 进行临床评估
- 准备技术文件
- 请求指定机构参与(对于 Is、Im 和 Ir)
- 准备使用说明和标签
- 检查是否符合第 10 条规定的制造商一般义务
- 欧盟符合性声明的抽签
- 贴上 CE 标志
将I类医疗器械投放欧盟市场后的步骤:
- 收集和评估上市后监控数据
- 警戒系统
- 不合格产品——如果制造商发现他们投放市场或投入使用的设备不符合欧盟 MDR,他们将立即采取必要的纠正措施使该设备符合要求,将其撤回或酌情召回。
顾问在 Class I (s/m/r) CE 标志中的作用
要想取得成功,必须有在欧盟 Is/Im/Ir 类合规性和临床评估方面有过经验的专家在场!海外顾问帮的作用如下:
- 指导和技术文件准备
- 确定测试要求并审查外部报告
- 根据 Meddev 2.7/1 Rev 4 准备临床评估报告。
- 安排公告机构并与他们协调直至颁发 CE 证书
- 安排欧盟代表从欧盟
- 安排欧盟的自由销售证书
以上就是关于“欧盟I类医疗器械符合性声明” 的相关内容,如果您想了解更多信息或办理医疗器械CE认证,请咨询在线客服或致电:400-106-2206。












