

如果你从来没有准备过医疗器械FDA上市前的通知,也就是通常所说的510(k)申请,弄清楚从哪里开始可能会让人望而生畏。虽然FDA的网站提供了一个信息金矿,但是要提取这些金块需要大量的挖掘。在这篇文章中,我们将提供一个如何接近fda510(k)批准程序的入门知识,解释该过程是如何工作的,并更多地讨论谓词设备研究和识别正确的产品代码。

医疗器械上市前通知510(k)流程的基本步骤
- 确认您的医疗设备的分类以及它是否属于510(k)途径。
- 使用FDA网站,为您的设备标识适当的三个字母的产品代码和法规编号。
- 在FDA数据库上进行研究,并选择要比较的谓词。
- 在FDA网站上搜索适用的FDA指导文件。
- 确定哪些国际“共识标准”可能适用于您的设备。
- 识别设备可能需要的临床数据和/或测试。
- 完成性能测试并进行临床研究(如果需要)。
- 将所有文档组装到510(k)应用程序中。
- 查看拒绝接受(RTA)清单,以确保您遵循FDA完整性指南。
- 支付510(k)审查费,获得收据,然后将510(k)提交给FDA。
- 在2周内收到FDA的确认,确认您的510(k)已接受实质审查。
- 如果确定您的510(k)基本等同,您将收到一封信函,并将其发布在FDA网站上,这将证明您的设备可以在美国合法销售。将不会颁发证书。
510(k)的正式名称是上市前通知。我们应该注意,FDA实际上并没有“批准” 510(k)申请,而是“批准”(授权)了要在美国销售的设备。这就是为什么510(k)被称为售前通知而不是售前批准(PMA)的原因,后者仅适用于III类设备。但是为了清楚起见,我们在这篇介绍性文章中使用了“批准”一词。FDA通过PMA流程“批准”了III类医疗设备。
1. 确认您拥有受FDA监管且需要510(k)的医疗设备
这看起来似乎很明显,但是第一步是要确认您的产品是受管制的医疗设备,并且需要通过510(k)批准流程。某些产品(即低风险设备I类产品)确实需要在FDA注册,但不需要经过FDA 510(k)流程。几乎所有II类设备都必须购买510(k)。以下是有关如何确定您的设备是否受到监管并需要510(k)的信息。
2. 确定适合您的医疗设备的产品代码和法规编号
FDA使用基于谓词的审查方法。这意味着,当您向FDA提交申请时,您将把您的医疗设备与FDA已经批准(谓词)的非常相似的设备进行比较。此过程与欧洲或加拿大使用的方法完全不同-这些市场对设备注册采用基于风险的规则方法,这意味着设备在安全性和性能方面必须在很大程度上依靠自己的优点。由于FDA要求您识别单个谓词设备,因此第一步就是找到一个。您可能已经知道哪种竞争产品可以为510(k)中的比较提供合适的谓词。无论如何,您应该使用FDA产品分类数据库开始研究。
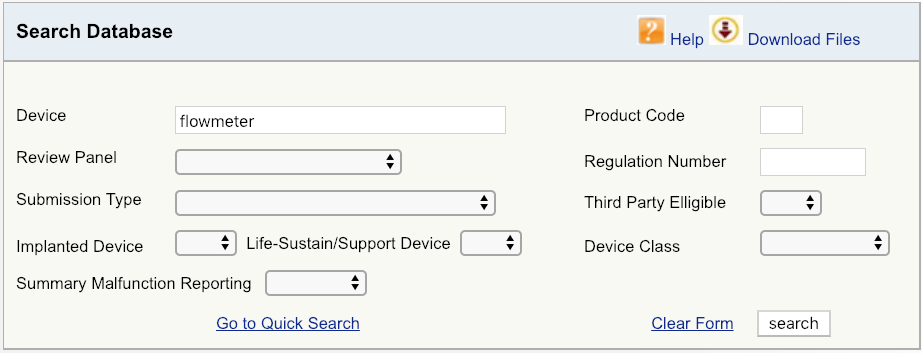
例如,假设您的公司正在向美国市场推出新型心血管血流仪。第一步将首先在FDA数据库上进行简单的设备搜索,如图所示,然后查看可用的选项。从产品的最广义定义开始-在这种情况下,仅是术语“流量计”。结果表明,与流量计相关的产品有六个独特的FDA产品代码。
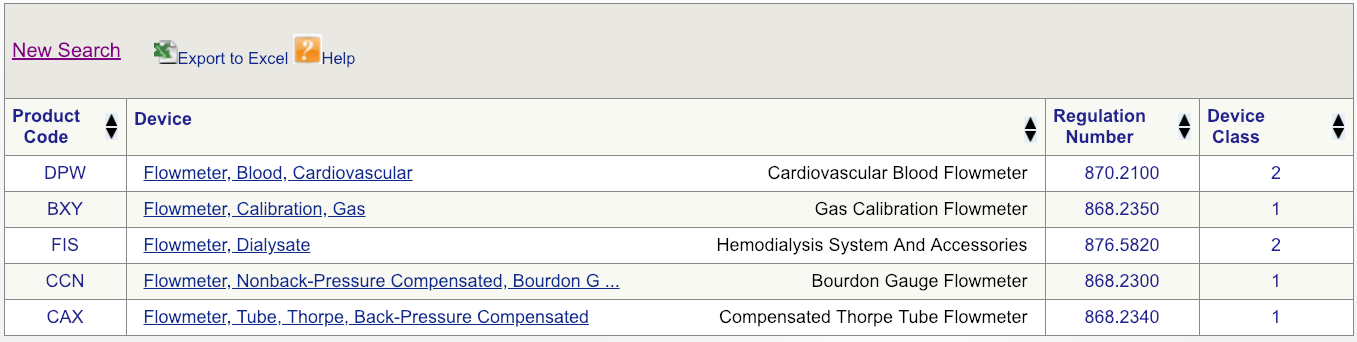
代码DPW看起来是最匹配的,但是要确保单击规则编号并仔细阅读说明。
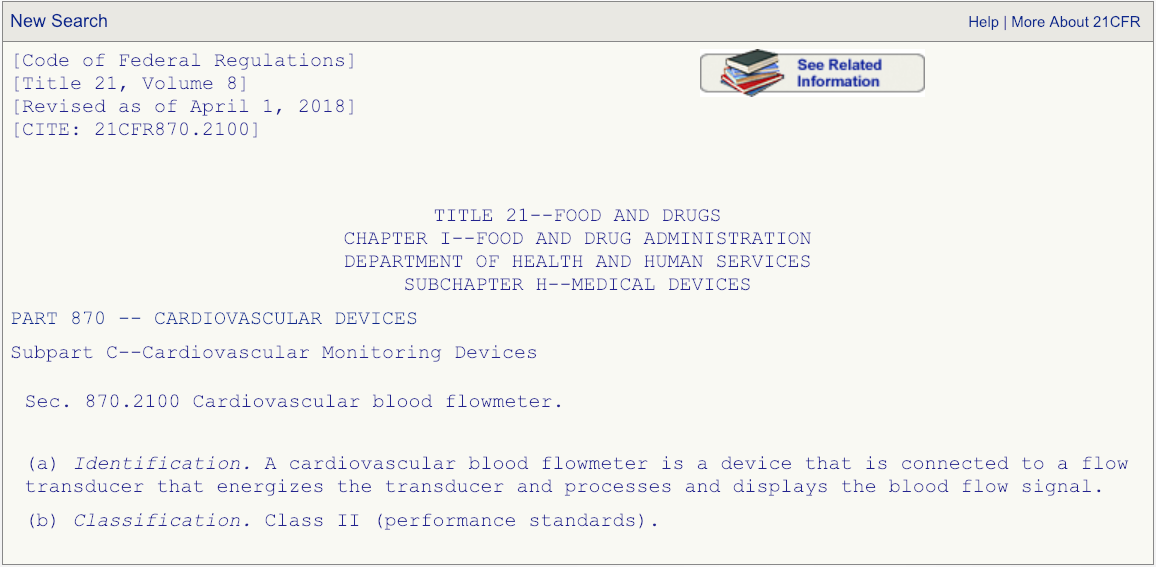
3. 为您的510(k)提交选择正确的谓词设备
在您阅读了与法规编号相关的描述之后,并完全确定产品代码DPW是正确的,然后进入FDA的510(k)数据库并搜索以产品代码DPW清除的所有设备。
这是棘手的地方,您需要小心。在此示例中,分类产品代码DPW下有131个已清除的医疗设备。哪一个最适合您的设备谓词?好吧,这里有一条建议:在检查选项时(希望您没有131个选项),最好按“ Decision Date”(决策日期)列进行排序,并从最近清除的设备开始。为什么?尽管可能会选择使用较旧的设备作为比较谓词,但FDA拒绝使用10年前清除的设备。
下一步将是单击每个设备的“摘要”链接,如图所示(请参见下面的示例页面)。
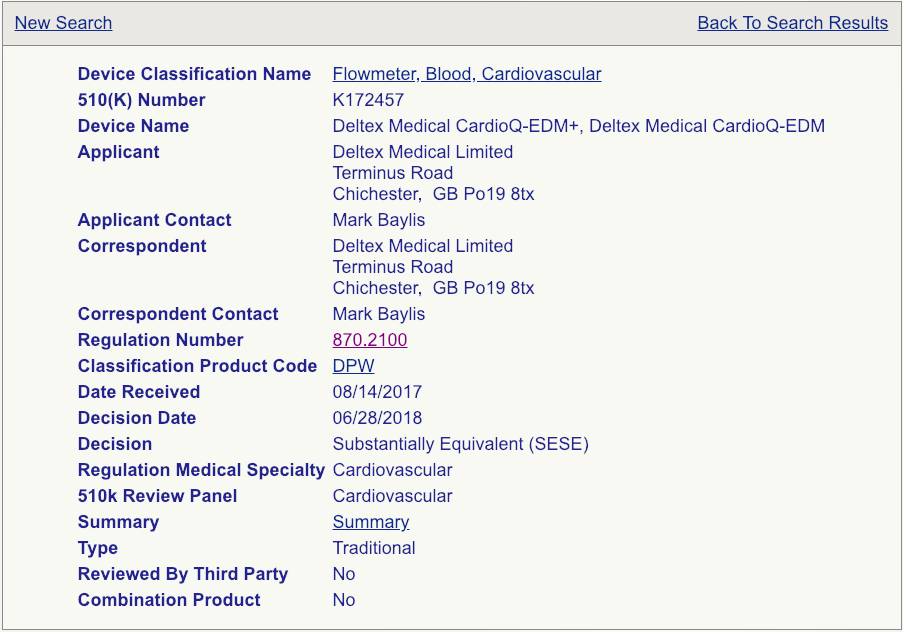
非常非常仔细地阅读这些摘要。注意预期用途,允许使用的适应症,进行的测试以及可能进行的临床研究。某些510(k)摘要提供的信息要多于其他摘要,因此,如果您要查看大量摘要,请确保您复习尽可能多的内容并将其知识汇总到电子表格中。您选择的谓词不必与您的设备相同,但需要足够接近,以免引发其他安全性和有效性问题。所选谓词必须具有相同的预期用途和使用指示。这非常重要。如果使用的指示不同,则该设备将不是合适的谓词。技术功能应与您的设备紧密匹配。选择正确的谓词对于成功提交至关重要,并且,如果您对自己的选择有保留,则应寻求经验丰富的FDA顾问的建议。
4. 果您的设备真正具有创新性,会发生什么?
FDA谓词注册系统的局限性在于它不容易适应创新。过去,这就是为什么一些引入创新技术的公司选择首先将其设备引入欧洲市场的原因。如果您拥有真正的新技术,或者您的设备结合了两种现有技术,则可以通过提交513(k)信息请求,要求FDA对设备的分类和法规要求发表意见。一些制造没有合适谓词设备的创新型低风险医疗设备的公司可以通过从头开始流程。这使FDA可以为没有当前相关产品代码的产品分配I类或II类名称和产品代码/法规编号。
您也可以要求与FDA进行Q提交会议(Q Sub),以澄清法规要求,就要包括的技术文件获得建议或讨论支持您提交的临床研究。这些简短的会议(亲自,通过电话或书面会议)使您可以提出一些实质性问题(取决于允许的时间),并且是确保您不浪费时间和金钱的一种有价值的方式。除非您对所要走的路和提交的内容完全有信心,否则我们建议您参加Q-Sub会议。(我们可以为您提供帮助。)由于FDA审评人员对附加信息(AI)的要求,将近三分之二的FDA 510(k)提交速度减慢了。
【海外顾问帮】是协助国内企业和个人跨境发展一站式的服务中心,协同全球专家顾问,坚持透明服务,打破跨境壁垒,为FDA 510(k)认证提供一站式服务,咨询电话: 400-106-2206。












