

ISO 13485:2016 是医疗器械制造商和供应商的质量管理体系的自愿性标准,在全球范围内用于开发和维护满足医疗器械市场需求的系统。修改ISO 13485的主要原因之一是使国际标准与自2003年以来不断发展的通用监管理念保持一致。ISO13485受到了世界领先的医疗器械监管机构的影响,例如FDA(食品和药品管理局)。

自2003年以来,医疗器械监管流程取得了长足进步。2016年,对ISO 13485的修订导致对产品安全,产品和流程风险管理要求的提高以及对监管机构的报告系统的改进。帮助用户满足通用法规要求。
ISO 13485:2016与FDA 21 CFR Part 820之间的关系
下图描绘了ISO 13485:2016如何帮助组织维持有效的质量管理体系,以满足适用的法规要求。最新的ISO 13485标准在其要求内还包含了常见的法规概念。基于此标准的组织可以逐步遵循FDA 21联邦法规(CFR)第820部分(质量体系法规)。根据对本法规的遵守情况,组织可以在美国商业销售医疗器械。第820部分定义了满足FDA法规的质量体系要求,被称为当前的良好生产规范。就要求而言,它更类似于ISO 13485。其他部分包括(例如)第810部分(专门处理医疗设备的召回程序)和第830部分(处理医疗设备的唯一设备标识)。
FDA 21 CFR Part 820和ISO 13485之间的异同
根据ISO 13485和FDA 21 CFR第820部分的目的,历史,范围以及相互之间的影响进行比较。下面的比较矩阵将帮助您了解标准和法规的工作范围,应用和领域。
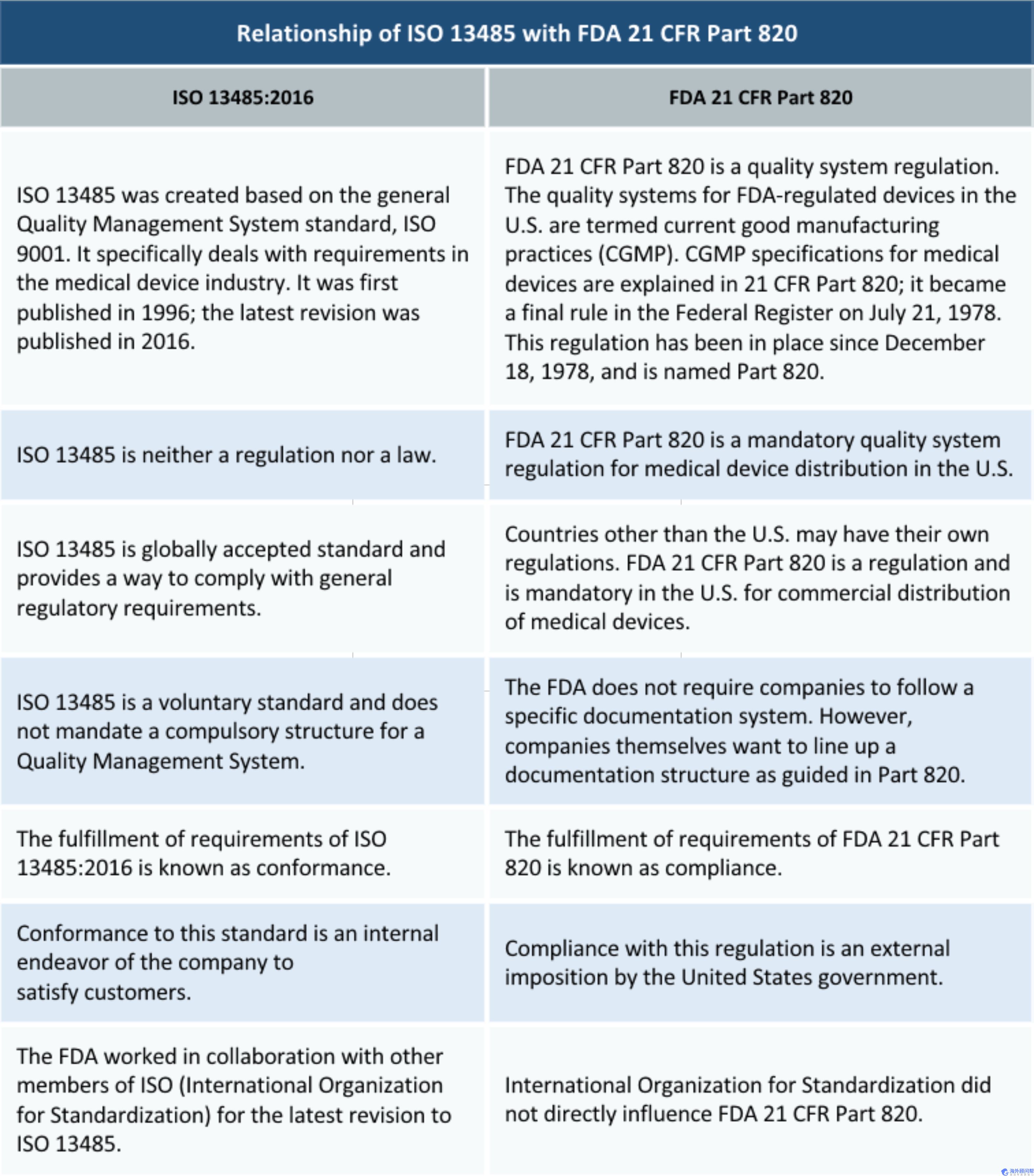
FDA的质量体系法规第820部分比ISO 9001:2015在更大程度上与ISO 13485:2016保持一致。许多国家在规范医疗设备方面均依赖ISO 13485:2016。FDA在ISO 13485:2016的修订中扮演了自己的角色,因为如果不同国家的需求相同,则行业更容易开发质量管理体系。由于要求相似,因此FDA和其他国家的设备监管机构可以更轻松地使用和讨论检查报告。
制造商可以使用ISO 13485:2016来符合FDA 21 CFR Part 820
因为FDA在ISO 13485的修订中起了重要作用,所以大部分820部分的法规要求都已包含在ISO 13485中。但是,有些要求可能未明确包含在ISO 13485中,例如设备历史记录(FDA 820.184部分) )。但是,ISO 13485标准的记录控制(条款4.2.5),产品实现计划(条款7.1)和标识(条款7.5.8)与设备历史记录的要求隐含相关。因此,顾问将对您当前的系统(根据ISO 13485开发)进行差距分析,然后提出一些在系统内采取的其他措施,以确保符合FDA 21 CFR Part 820。
ISO 13485为制造商和供应商提供了满足全球通用法规要求的框架,并为满足FDA Part 820要求以及全球其他法规机构的要求奠定了坚实的基础。
【海外顾问帮】是协助国内企业和个人跨境发展一站式的服务中心,协同全球专家顾问,坚持透明服务,打破跨境壁垒,为FDA认证、ISO 13485认证提供一站式服务,咨询电话: 400-106-2206。












