

根据欧盟MDR对医疗器械进行分类
医疗器械制造商过渡到遵守新法规的过程中会感觉到的欧盟MDR的主要变化之一是对器械分类要求的改变。随着医疗器械分类的变化,对制造商的要求也随之变化。设备分类的更改也将影响医疗设备制造商与其公告机构互动的方式和时间。所有这些都是至关重要的,因为它会影响设备获得CE标记的能力,这是在欧洲合法销售您的设备所必需的。
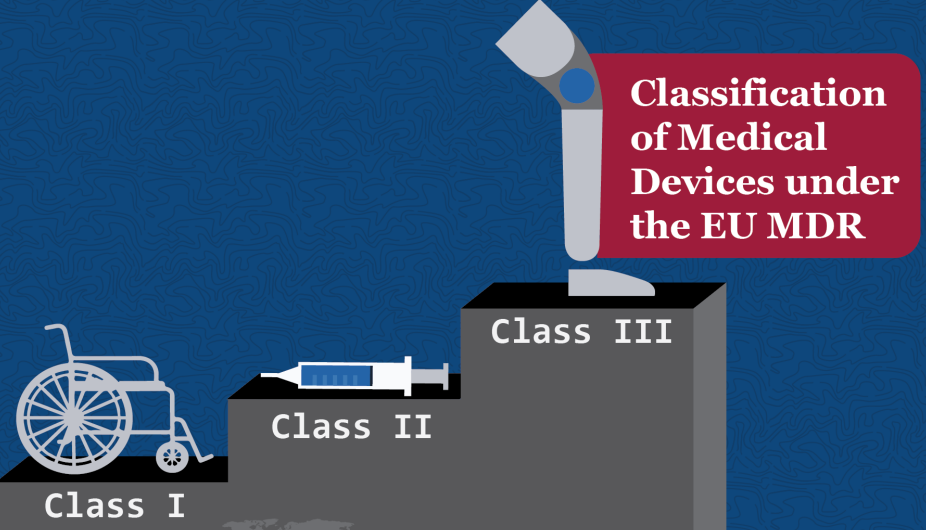
当前的医疗设备指令(MDD)和MDR都将医疗设备分为以下类别之一:
- I类(低风险)
- IIa类(中等风险)
- IIb类(中/高风险)
- III类(高风险)
与FDA一样,EU法规采用基于风险的方法对医疗设备进行分类。[1]您的医疗设备风险越高,您必须遵守的法规越多。根据MDD,该指令的附录IX中有18条分类规则。在即将到来的MDR中,有22种分类规则,部分是由于范围更广。MDR将适用于某些未通过MDD管制的产品,例如无预期医疗目的的设备,例如非矫正(装饰性)隐形眼镜。2MDR还将特别管制结合了纳米材料的设备和用非活人体组织制造的设备,目前已从MDD中豁免。
新法规还带来了与某些设备相关的分类更改。所有有源植入式设备及其配件将被视为III类。打算通过体孔口使用或应用于皮肤的任何基于物质的设备可能都不属于I类,因此,当前属于I类的任何基于物质的设备将通过新法规进行上级分类。任何使用MDR进行分类的设备的制造商必须遵守更严格的要求,并且可能需要更多地与他们的公告机构进行接触。
此外,医疗应用行业也将受到MDR重新分类的影响。目前,欧洲大多数医疗软件都属于I类,但MDR对医疗设备软件的要求将会更加严格。3医疗设备软件开发人员现在必须遵守更严格的规则,才能继续在欧盟销售其软件。
海外顾问帮可以帮助您从MDD过渡到即将到来的欧盟MDR,请通过在线客服或拨打400-106-2206与我们联系,以了解我们如何提供帮助!












