

随着欧盟MDR过渡期限越来越近,医疗器械企业从现在开始有必要重视MDR标签要求,因为标签标注不精确或者是不符合要求都有可能造成公司无法继续在欧洲销售其产品,从而造成大量库存积压,甚至引发成本高昂且损害公司声誉的产品召回事件。那么,欧盟医疗器械法规MDR标签要求有哪些呢?

1. MDR法规要求
先来看一下MDR法规的具体要求:

法规附录I中第III章23.2对于产品标签要求必须注明:
(a) 器械的名称或商品名称;
(b) 使用者识别器械所必需的详细信息、包装内容以及对于使用者不明显的器械预期用途;
(c) 制造商的名称、注册商号或注册商标及其注册营业地点的地址;
(d) 授权代表的姓名和授权代表的注册营业地点地址(若制造商在欧盟以外有其注册营业地点);
(e) 若没有指明可安全使用的日期,则指明制造日期。若日期清晰可辨,制造日期可作为批号或序列号的一部分。
(f) 指明适用的任何特殊储存和/或处理条件;
(g) 若以无菌方式提供器械,还应指示其无菌状态和灭菌方法;
(h) 需要立即引起器械使用者和任何其他人的注意、需要采取的警戒或预防措施。该信息可保持最小量,在这种情况下,更详细的信息将出现在使用说明中,同时考虑到预期使用者;
(i) 若器械用于一次性使用,则相应指明。制造商的一次性使用指示应在整个欧盟内保持一致;
(j) UDI 载体应添加在该器械标签和所有更大包装上;
(k) 标签应明显、清晰和不可磨灭地添加在器械或其无菌包装上。考虑到器械性质,无法或不适合将标签添加到器械上时,应将 CE 标识添加在包装上。CE 标识也应加贴在有使用说明和任何销售包装中;
(l) 应采用器械上市国(同时也是成员国)指定的欧盟官方语言编写,也可以采用预销往国的当地语言;
(m) 标签上所需的信息应在器械本身上提供。若不可行或不适当,则某些或所有信息可显示在各单元的包装上和/或多个器械的包装上。
2. MDR标识举例
那么到底哪些标识是在标签(包装)上必须体现的呢?
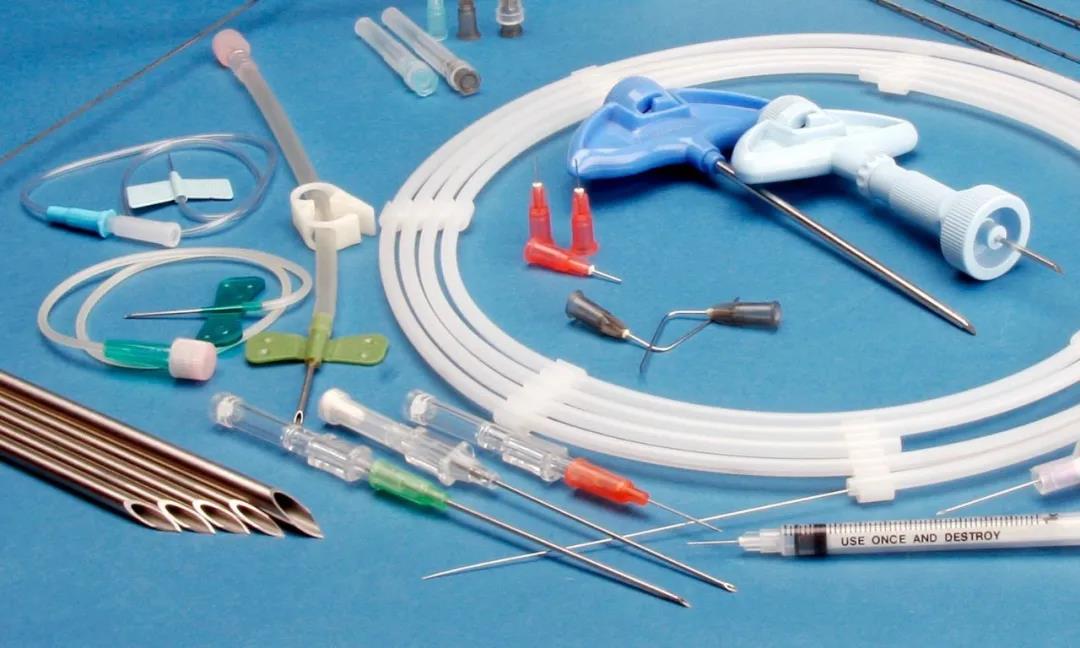
常用无源器械标识
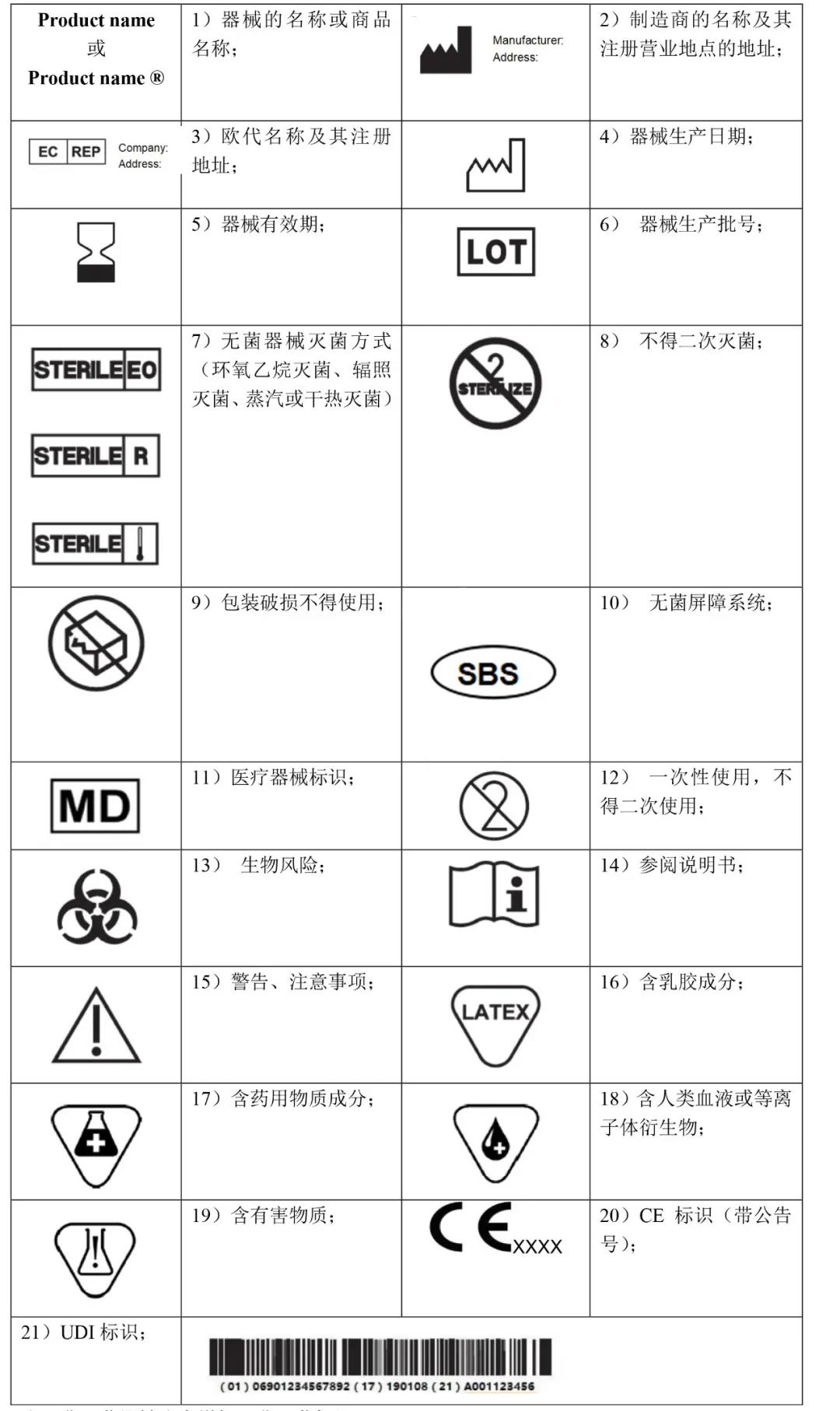
注:非灭菌器械注意增加“非灭菌标识”。
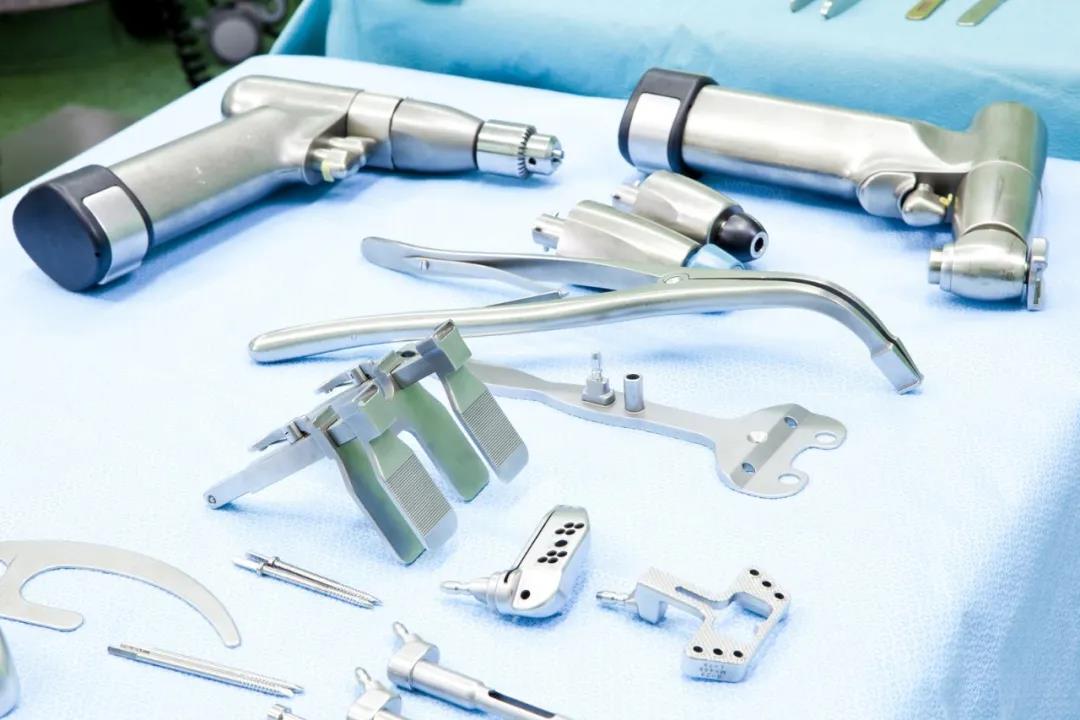
常用有源器械标识
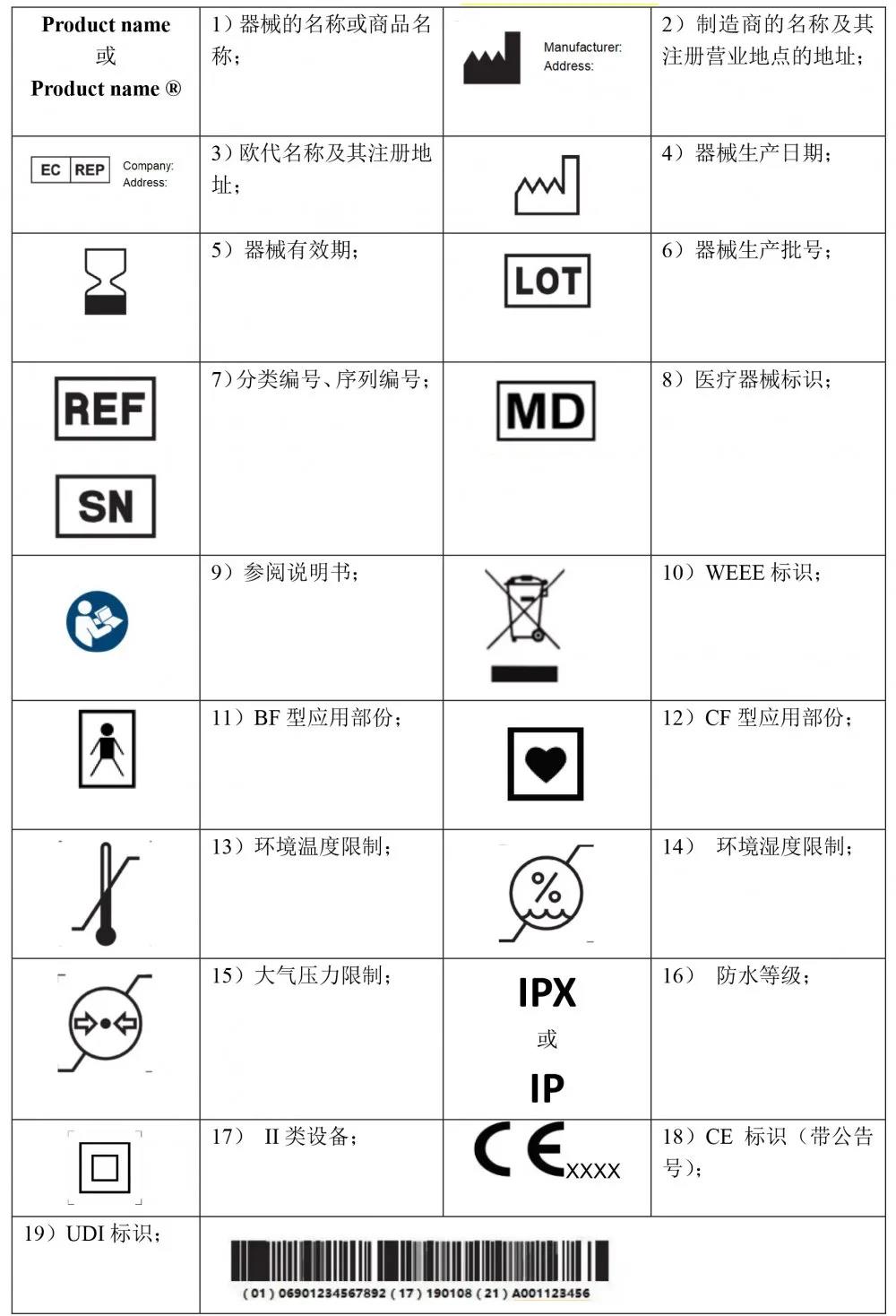
注:有源器械涉及的标识很多,需根据器械的专标要求进行补充。
常用的包装标识
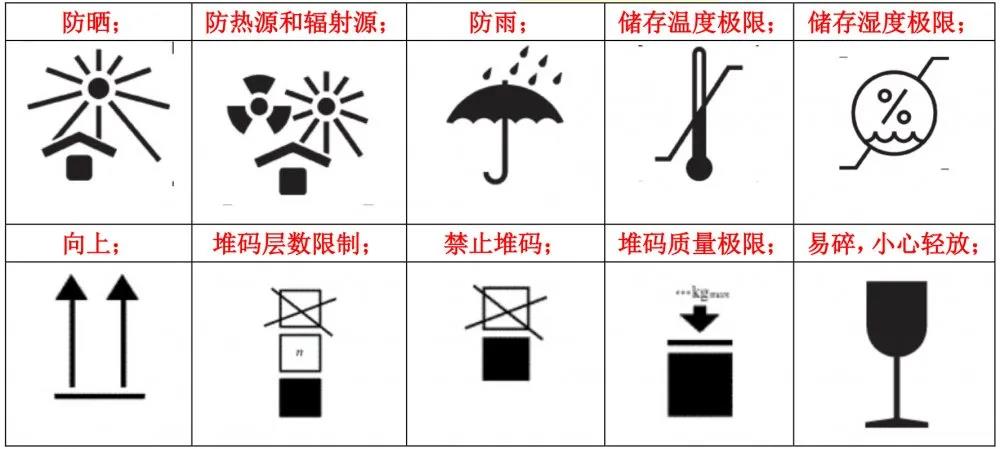
注:制造商信息、欧代信息和外包装UDI可以体现在外包装上。
3. MDR标签信息更改
企业如果想变更标签上的信息,比如器械名称或商品名称、规格型号、制造商名称和地址、欧代名称和地址、器械有效期、灭菌方式发生改变时,应及时通知公告机构,经评审确认无误后方可使用新标签。擅自使用不符合要求的标签会有被勒令召回的风险(MDR一类器械除外)。虽然MDR一类器械没有公告机构评审,但设计标签时仍需满足MDR法规附录I 第23.2条的要求。
为应对未来潜在的法规变化,制造商需提前规范并优化标签流程,这也是上市前合规战略计划中的重要一项准备工作!
以上就是关于“欧盟医疗器械法规MDR标签要求”的相关内容,了解更多点击咨询。












