

欧盟医疗器械MDR法规介绍
实际的医疗器械指令(MDD)93/42 / EEC [ 1 ]和有源植入式医疗器械指令(AIMDD)90/385 / ECC [ 2 ]是欧洲各种医疗器械的基本指令。这些指令对所有成员国都是强制性的,必须由国会在给定的时限内纳入国家立法。不允许减少或更改该指令的要求,但是议会可以执行其他要求。
与医疗器械指令相反,医疗器械法规(MDR)[ 3 ]直接来自布鲁塞尔的欧洲委员会,未经国家议会的任何批准,必须在给定的期限内作为欧洲超国家法律适用。各国议会可以解决其他国家要求。
新型MDR的原因之一是PIP丑闻:一家制造商使用便宜的工业硅用于乳房植入物,而不是超纯医用硅。一旦这个丑闻被公之于众,委员会决定收紧该指令,以防止这种刑事诉讼。
新的MDR的原因之一是PIP丑闻:一家制造商使用了便宜的工业硅来代替超纯的医用硅。一旦这一丑闻被公之于众,委员会决定加强指令,以防止这种犯罪过程。
欧盟医疗器械MDR法规变化
实际的MDD 93/42 / EEC有60页,包括23条(24页)和12个附件(36页)。新的MDR包括333页和123条(92页)和17个附件(241页)。MDR的主要更改或新内容是:
- 制造商可以任命公司中具有医疗器械领域专业知识的合格人员。
- 欧洲EUDAMED数据库将得到广泛扩展。
- 指定机构的活动和审核证书将在欧洲范围内统一(MDR证书)
- 合格评定程序将有所变化。与MDD附件VI相类似的程序不再可用。
- 实施了所谓的审查程序:指定机构可以承诺将任何新的高风险产品合格性评估申请报告给决定产品临床评估的医疗器械协调小组(MDCG)。
- 一些产品的分类将发生变化:现在属于IIb类产品(MDD)的某些可植入产品将满足III类产品的要求,而软件很少会成为I类产品。
- 对一次性产品进行再加工的要求增加了。
- 医疗器械技术文档的更详细规范包含在MDR的附件II中。MDR的新要求是不断更新文档。
- 技术文档的保留期限从MDD的5年增加到MDR的10年增加了一倍。
- 每个医疗产品都需要一个唯一的设备标识(UDI)。借助这种识别系统,委员会计划对医疗产品进行识别和注册。
- 对医疗器械贴标签有新的要求。
- 将对临床评估和临床研究进行更详细的规定。
- 必须使用现在重要的上市后监督的上市后信息来更新临床评估。
下图显示了MDR下文档之间的依赖关系:
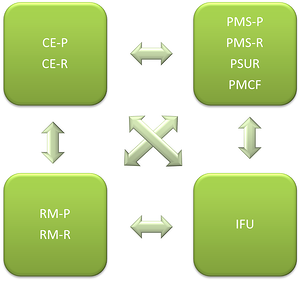
CE-P:临床评估计划 CE-R:临床评估报告
PMS-P:上市后监视计划 PMS-R:上市后监视报告
SUR:定期安全更新报告 PMCF:上市后临床随
IFU:使用说明
RM-P:风险管理计划 RM-R:风险管理报告
新MDR的目的是加强MDD,以使医疗产品更安全,更可靠。通过将现有指令升级为欧洲法规,合格评定程序将在所有成员国中成为统一的过程,从而使结果一致且更加透明。为此,MDR将更加强调医疗产品制造商的文档。这将导致符合MDR的最终医疗产品的成本上升。由于必须对已经在MDD下批准的医疗产品制定新的合格评定程序,承担上市后监督和上市后临床随访以及合规官的额外工作,因此将产生额外费用。对于中小型企业,存在风险,即他们没有必要的人力和/或财力来遵守新法规。结果,存在许多中小企业及其产品从市场上消失的风险。
以上就是关于“什么是欧盟医疗器械MDR法规”的相关内容,了解更多点击咨询。
【海外顾问帮】是协助国内企业和个人跨境发展一站式的服务中心,协同全球专家顾问,坚持透明服务,打破跨境壁垒,为医疗产品CE认证提供一站式服务,咨询电话:400-106-2206。












